| Pérdida de tolerancia a antígenos propios | Lámina 44 |
||||
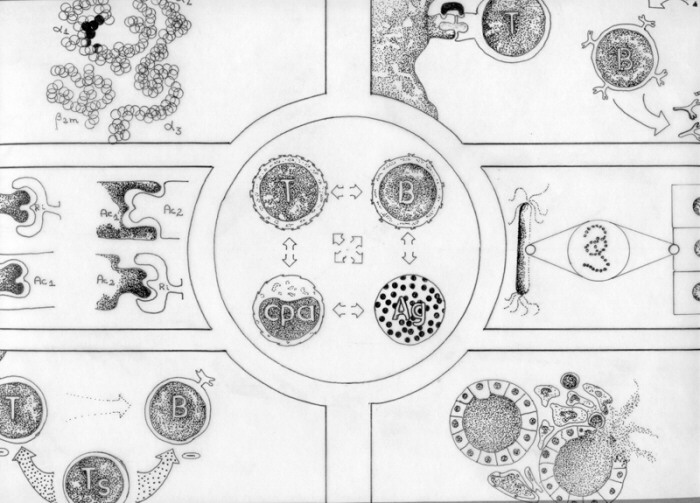
Las Enfermedades Autoinmunes surgen como consecuencia de una pérdida de la tolerancia a antígenos propios. Como se ha dicho, el tipo de respuesta inmune, inmunogénica o de tolerancia, depende básicamente de la calidad del antígeno y de las interacciones entre células presentadoras de antígeno, linfocitos T y linfocitos B (figura central). Los mecanismos de pérdida de la tolerancia a antigenos propios que han sido descritos son complejos y en gran medida especulativos. En general, la ausencia de tolerancia a antigenos propios puede deberse a defectos intrínsecos del sistema inmune (1, 2 y 3) o bien a alteraciones relacionadas con el antígeno y su reconocimiento (4, 5 y 6)
Entre los primeros se ha descrito alteraciones en la presentación del antígeno por moléculas MHC durante la deleción clonal en el timo o en la periferia (1), en la regulación por anticuerpos antiidiotipo (2) y en la actividad supresora de los linfocitos T. Los principales mecanismos relacionados con el antígeno,
incluyen la activación policlonal de células
B por antígenos bacterianos y de células T
por superantígenos (4) el mimetismo molecular y reacciones
cruzadas (5), y la liberación de antígenos
secuestrados (6). 1.Alteraciones en la presentación de antígeno por las moléculas codificadas por el MHC: Se ha postulado que la susceptibilidad a padecer enfermedades autoinmunes estaría más bien ligada a genes del sistema mayor de histocompatibilidad que a aquellos que codifican el TCR. Algunas enfermedades autoinmunes se relacionan con genes que codifican las moléculas MHC clase I (Espondilitis anquilosante y Psoriasis) y otras con alelos del MHC clase II ( Lupus Eritematoso Sistémico y Poliarteritis Nodosa). La asociación con estos alelos es multifactorial, esto es, con una combinación de alelos y no con uno de ellos en particular. Por ejemplo, los individuos que heredan las especificidades definidas serológicamente HLA A1, B8 y DR3 tienen mayor suceptibilidad de padecer Diabetes tipo I, Miastenia Gravis y Lupus Eritematoso Sistémico. La presencia de estos alelos determinaría una forma particular de apareamiento de las cadenas alfa y beta del MHC que influye en el reconocimiento por parte del TCR del complejo MHC-antígeno propio. Otros autores han postulado que la mayor susceptibilidad que presentan las personas con estos alelos se debe a cierto grado de homología con algunos virus. Así por ejemplo, al comparar la estructura del DQ beta de estos alelos con la de genomas virales, se ha visto que el genoma del virus Epstein Barr y de la Rubeola tienen secuencias de alta homología con el DQ 3.2. Este alelo está presente en el 65% de los pacientes con Diabetes tipo I. De esta manera, una infección viral podría originar respuestas de anticuerpos que reaccionan con moléculas MHC propias. La existencia de estas asociaciones entre ciertos alelos MHC y EAI esta siendo intensamente estudiada, sin embargo, el mecanismo involucrado en el inicio y persistencia de EAI no está aún claramente establecido. La presencia de determinados alelos MHC podría influir en la presentación de antígenos propios durante la a) la deleción clonal o b) durante la respuesta inmune. a) Durante la deleción clonal: utilizando técnicas tales como cultivo de células que conforman microambientes tímicos y anticuerpos monoclonales, ratones transgénicos y ratones SCID (incapaces de expresar TCR) se ha avanzado bastante en la comprensión de los mecanismos involucrados en la deleción clonal. Los ratones transgénicos poseen material genético introducido artificialmente. Al transferir genes que codifican TCR de una especificidad determinada a huevos fertilizados de ratón, se obtiene animales que expresan ese TCR entre otros. Si se usa como receptres de estos genes a ratones SCID ( que padecen de inmunodeficiencia combinada severa y no expresan TCR ), se obtiene animales transgénicos que expresan una sóla especificidad en todos los TCR de sus linfocitos T. Utilizando anticuerpos monoclonales, se puede seguir el destino de estos linfocitos durante su maduración y evaluar los diferentes mecanismos involucrados en la deleción clonal. Mediante estas técnicas se ha podido postular que las células T inmaduras estarían programadas para morir a menos que sean estimuladas adecuadamente por la interacción de su TCR con el complejo MHC-Ag. Se cree que si esta interacción es de alta afinidad o carece de señales coestimuladoras, las células sufren apoptosis ( muerte programada). La deleción clonal de clones autorreactivos podría ser evitada por lo tanto afectando la avidez de los TCR por su ligando o alterando las señales coestimuladoras entre otros mecanismos. En todo caso, estas y otras ideas son aún hipotéticas y se puede concluir que los mecanismos que dan cuenta de alteraciones en la deleción clonal son desconocidos. b) Durante la presentación de antígenos en la periferia: se ha descrito la presencia de alelos MHC con secuencias aminoacídicas en su región hipervariable ( cleft ) que tienen especial afinidad por autoantígenos en los individuos suceptibles. En relación con este tópico, se ha señalado también que citoquinas generadas en inflamaciones de diversa índole, promueven la expresión de moléculas MHC clase II en células que normalmente no lo expresan aumentando la probabilidad de activar linfocitos autorreactivos y generar respuestas autoinmunes. 2. Anticuerpos anti-idiotipo: 3. Linfocitos T supresores: Los linfocitos T CD8+ son capaces, según algunos autores, de generar una actividad supresora mediante la liberación de productos específicos solubles o de algunas citoquinas (IL-10 e IFN gamma). La existencia de linfocitos T supresores se apoya en la posibilidad de transferir adoptivamente la tolerancia a diversos antígenos de un animal experimental a otro. Estos linfocitos parecen tener importancia en la génesis de enfermedades autoinmunes órgano-específicas, ya que serían los encargados de mantener tolerantes a los clones autorreactivos que no fueron deletados por encontrarse su antígeno en lugares anatómicos no accequibles para el sistema.
B. Alteraciones relacionadas con el antígeno y su reconocimiento: 4. Activación policlonal de linfocitos T y B: Los linfocitos T pueden sufrir activación policlonal por moléculas que han sido denominadas superantígenos. Estos, que pueden ser endógenos (Mls) o exógenos (SEB o enterotoxina estafilocócica B) activan a linfocitos T uniéndose a la cara lateral del complejo TCR-MHC siempre y cuando el TCR presente un determinado alelo en la cadena beta. Esta unión, si ocurre en el timo durante la ontogenia, se traduce en deleción clonal. Cuando se produce en la periferia en un sistema inmune maduro, se traduce en la activación de todos los linfocitos que comparten ese alelo. El papel que podrían jugar estos superantígenos en la génesis de enfermedades autoinmunes está siendo evaluado. En el caso de la activación policlonal de linfocitos B, las respuestas se producen por acción de endotoxinas bacterianas y virus Epstein-Barr principalmente. Estas respuestas se manifiestan como la presencia de anticuerpos con múltiples especificidades que reconocen una gran variedad de antígenos, tanto propios como ajenos. Entre ellos, se ha encontrado autoanticuerpos anti- DNA, anti- tiroglobulina y anti-insulina. Ultimamente se ha descrito que estas respuestas policlonales estarían a cargo de una subpoblación de linfocitos B CD5+ que conforman un 20% de los linfocitos B periféricos. Se ha encontrado un aumento de estas células en pacientes y animales experimentales con diversas enfermedades autoinmunes. 5. Mimetismo molecular y reacciones cruzadas: El mimetismo molecular y las reacciones cruzadas serían, según muchos autores, los principales mecanismos responsables de la pérdida de la tolerancia a antígenos propios. Existen regiones de alta homología entre moléculas de virus, bacterias y parásitos con moléculas MHC y otras presentes en nuestra especie. Las enfermedades autoinmunitarias podrían ser provocadas cuando los epitopos microbianos son lo suficientemente similares a los propios como para originar reacciones cruzadas. Por ejemplo, en los pacientes con Espondilitis Anquilosante se ha visto una alta incidencia del alelo MHC-I B27, el cual comparte en su secuencia aminoacídica 5 a 6 aminoácidos con la nitrogenasa de la Klebsiella neumoniae. Se ha postulado, que una vez que estos microorganismos provocan daño tisular en individuos susceptibles, la injuria inicial expone permanentemente antígenos propios que presentan reacción cruzada con los anticuerpos y perpetúan el daño. Este mecanismo operaría en diversas enfermedades infecciosas tales como lepra, TBC, infecciones por estreptococos las que se asocian a enfermedades autoinmunes. Asimismo, la Diabetes tipo I se asocia con infecciones virales. 6. Liberación de antígenos secuestrados: Finalmente, otra manera de romper la tolerancia a antígenos propios e inducir enfermedades autoinmunes es por medio de la liberación de " antígenos secuestrados ". Estos antígenos, por su ubicación anatómica no han tomado contacto con el sistema inmune y por lo tanto no han inducido tolerancia a ellos. Si por algún traumatismo o infección se liberan masivamente, existirá una respuesta inmune frente a ellos que podrá causar o perpetuar el daño tisular. Se puede concluir entonces, que las enfermedades autoinmunitarias surgen a raíz de una pérdida de la tolerancia a antígenos propios por los mecanismos (aún hipotéticos) mencionados,lo que se traduce en la activación de clones linfocitarios autorreactivos. Las consecuentes respuestas efectoras, producen daño tisular órgano-específico o sistémico a través de los mecanismos de daño inmunológico tipos II,III y IV. Existen cerca de cuarenta Enfermedades autoinmunes diferentes que afectan entre un 5 y un 7% de la población. Considerando que los individuos presentan normalmente un número importante de células potencialmente autoagresivas y que todos los individuos sanos pueden manifestar respuestas autorreactivas, es necesario explicarse porqué algunos de ellos desarrollan Enfermedades autoinmunes. Hasta el momento el problema sigue siendo una gran incógnita si bien hay avances importantes en relación a factores genéticos que determinarían una predisposición a padecer estas enfermedades. |
|||||