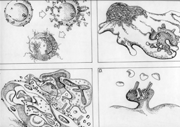
Mecanismo
de daño tipo II o citotóxico
|
|
y
|
|
|
Haz click sobre cada
lámina para ver detalles
|
Este mecanismo de daño
es responsable de algunos cuadros de hipersensibilidad.
Sin embargo, su participación es más importante
en autoinmunidad.
Los antígenos involucrados en hipersensibilidad son
sustancias exógenas solubles o de origen bacteriano
que se unen a a proteínas de membranas celulares
o basales. También pueden ser antígenos que
conforman los grupos sanguíneos en glóbulos
rojos como ocurre en las reacciones post-transfusionales
y en la enfermedad hemolítica del recién nacido.
En autoinmunidad, los antígenos son componentes propios
o endógenos que forman parte de membranas. Finalmente,
el daño mediado por este mecanismo puede surgir a
raíz de reacciones cruzadas entre antígenos
propios y bacterianos.
Las inmunoglobulinas que participan son la IgG y la IgM
producto de respuestas primarias o secundarias ante la presencia
de los antígenos mencionados. En algunos casos hay
también participacion del complemento.
El mecanismo de daño tipo II se presenta en cuatro
modalidades:
a. Lisis por complemento.
El antígeno o hapteno unido a membrana o formando
parte de ésta, origina una respuesta humoral tradicional
de IgG e IgM. Estos anticuerpos se unen al antígeno
en la membrana celular y activan al complemento por vía
clásica. Se produce la lisis osmótica de la
célula cuando la activación llega hasta la
formación del Complejo de Ataque a Membrana (MAC).
Ejemplos en hipersensibilidad: anemia hemolítica
postransfusional por incompatibilidad ABO y algunos tipos
de hipersensibilidad a drogas. La primera, aunque no es
frecuente, es de graves consecuencias. La hemólisis
es intravascular, originando hipotensión, hemoglobinuria
o hemorragia difusa. Se puede llegar a un estado de shock,
coagulación vascular diseminada, insuficiencia renal
y a veces, la muerte.
La anemia hemolítica inducida por penicilina es un
ejemplo de este mecanismo de daño. Se requiere de
altos títulos de IgG dirigido contra el determinante
antigénico de la droga unido a la membrana celular
de los glóbulos rojos. La hemólisis se inicia
generalmente 7 días después de la primera
exposición a la droga o después de un tiempo
menor en individuos previamente sensibilizados a ella. Otras
drogas tales como la quinina, quinidina y nitrofurantoína
inducen anticuerpos IgG que se unen a antígenos de
ciertos grupos sanguíneos tales como los sistemas
Rh, Kidd, Kell y a algunas proteínas de membrana
de plaquetas originando trombocitopenia.
Ejemplos en autoinmunidad: neutropenia en el Lupus Eritematoso
Sistémico (LES) en la cual se producen auto-anticuerpos
contra estructuras de membrana de leucocitos.
b. Opsonización
por anticuerpos o complemento.
Las inmunoglobulinas y fragmentos C3b unidos a membrana,
producen la opsonización de la célula, la
cual es fagocitada por macrófagos en bazo, ganglios
linfáticos e hígado. Se produce de esta manera
un acortamiento de la vida media de las células afectadas.
Ejemplos de hipersensibilidad: algunos tipos de anemias,
leucopenias y trombocitopenias inducidas por drogas y anemia
hemolítica del recién nacido por incompatibilidad
Rh y ABO. La anemia hemolítica del recién
nacido por incompatibilidad Rh se debe a la acción
de anticuerpos maternos IgG anti D que atraviesan la placenta
uniéndose a los glóbulos rojos del feto, los
que son eliminados en el bazo. La anemia hemolítica
del recién nacido por incompatibilidad ABO es más
frecuente en madres de grupo 0 con hijos grupo A ya que
estas poseen isoaglutininas IgG naturales. Sin embargo,
dado que los antígenos A y B del feto no están
bien desarrollados, las manifestaciones no suelen revestir
mayor gravedad.
Ejemplos en autoinmunidad: anemia hemolítica autoinmune
presente en enfermos con Lupus Eritematoso Sistémico,
donde los autoanticuerpos son IgG anti grupo sanguíneo
Rh llamados anticuerpos calientes ya que interactúan
con los eritrocitos del paciente a la temperatura corporal.
c. Inflamación en
membranas basales.
Cuando el antígeno se une o forma parte de membranas
basales, la activación de la vía clásica
del complemento por las inmunoglobulinas unidas a ellos,
se traduce en una inflamación exudativa principalmente
por acción de las anafilatoxinas C3a y C5a. Estas
moléculas inducen la degranulación de células
cebadas y basófilos liberándose mediadores
químicos de la inflamación tales como histamina,
leucotrienos y prostaglandinas. Una biopsia del tejido afectado
incubada con anticuerpos anti-IgG o IgM fluorescentes, muestra
un patrón contínuo de fluorescencia.
So es frecuente encontrar cuadros de hipersensibilidad mediados
por esta variedad de mecanismo de daño tipo II. Como
ejemplo en autoinmunidad cabe mencionar al Sindrome de Goodpasture
que se produce a raíz de la presencia de autoanticuerpos
anti-membrana basal glomerular y pulmonar (IgG anti MBG).
Su etiología es desconocida, sin embargo se ha descrito
asociaciones con terapia con penicilamina, infección
por el virus A2 de la influenza y exposición a solventes
orgánicos. La biopsia renal y pulmonar muestra tinción
fluorescente continua cuando se incuba el tejido con anticuerpos
anti IgG y anti-complemento. La mayoría de los pacientes
son hombres jóvenes, los que presentan neumonía
hemorrágica y nefritis. Generalmente se inicia con
hemoptisis, tos y disnea y continúa al cabo de unas
semanas con nefritis productiva y muerte.
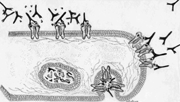
d. Estimulación
o interferencia con receptores.
En algunas afecciones autoinmunes, se producen anticuerpos
anti-receptores de membrana. Estos anticuerpos pueden estimular
receptores mimetizando la acción de la hormona (ejemplo:
Enfermedad de Basedow- Graves) o bien bloquearlos impidiendo
la unión del ligando correspondiente ( ejemplo: Miastenia
Gravis).
En la enfermedad de Basedow-Graves se produce un hipertiroidismo
con todas las consecuencias clínicas propias de esta
condición.
En la Miastenia Gravis se produce un debilitamiento progresivo
de la actividad muscular, al no poder la acetilcolina ejercer
su acción estimuladora en la placa neuromotora. Esto
se puede deber por una parte a que el anticuerpo anti receptor
bloquea la zona de fijación de acetilcolina en ellos.
Otro mecanismo posible es la formación de enlaces
cruzados entre receptores por los anticuerpos lo que se
traduce en endocitosis y degradación por lisosomas
de los complejos receptor-anticuerpo. Finalmente, la activación
del complemento hasta C3 puede acelerar la pérdida
de receptores y hasta C9 destruir la membrana post-sináptica.
Algunos autores incluyen también en este mecanismo
de daño inmunológico a la actividad ADCC de
células NK.